Data Analysis
Mascot Searching
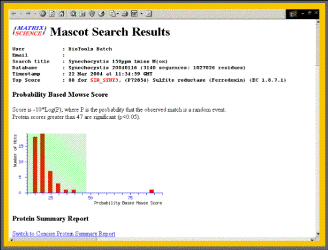
Mascot search result of a MALDI-MS mass list.
The spot had been picked from a 2D-high resolution NEPHGE gel. Sample: Synechocystis sp. 6803 thylakoides.The protein identified is Ferredoxin, a redox protein, that accepts electrons from photosystem I. It is located at the end of the photosynthetic electron transfer chain at the cytoplasmic side of the thylakoid membrane.
(Courtesy of Dr. Irrgang, Max-Volmer-Institut, Technische Universität Berlin, Germany)
MALDI-MS-Fingerprinting
A Maldi-MS instrument combining high mass accuracy & resolution, good throughput and a choice of targets which can address different samples quantities, is, together with the availability of complete genetic sequence information for the organism under investigation the main pre-requisite for successful Maldi-MS-Fingerprinting. If these pre-requisites are met and rigor analysis of the data obtained is applied, most proteins i.e. those which are not or only moderately posttranslationally modified, will be readily identified by this technique.
The first analytical challenge remaining is to actually realize a high mass accuracy for each individual signal of a mass list. <200 ppm can be achieved by external calibration, <10 ppm can be achieved by internal calibration. In both cases values actually reached will to some extend depend on the number and distribution of calibration masses.
Finding the signal/noise treshold which constitutes a good compromise between the loss of weak sample signals and the increase in the number of artifact signals as well as knowing and consequently removing all "contaminating" masses from the list prior to Mascot-Searching, are further steps needed to achieve success.
Finally, repeated database searching considering those protein modifying reactions the sample might most likely have undergone within the time span between transcription and impact on the mass spectrometers detector, is another factor that will improve the likelihood of identification by Mascot-Searching of Maldi-MS-fingerprints.
ESI-MS-Data
Success in Mascot-Searching for ESI-MS sequence data information relies on the length and specifity (uniqueness) of the sequences obtained. The amount of sample available determines the lenght of measuring time, i.e. how many different fragments might be investigated in MS/MS-mode. A high number of MS/MS-investigation increases the likelihood to obtain unique sequences. However, since measuring time (i.e. sample amount) usually is one limiting factor, a well optimized strategy to quickly identify the most promising candidate ions for MS/MS is needed.
Thorough exclusion of signals which are known to constitute autolytic fragments of the cleaving protease and other artifacts before MS/MS using exclusion lists significantly increases the likelihood to obtain specific information, although, some artifacts may still slip through this exclusion list due to unspecific cleavage (insource decay fragments).
The remaining lack of specificity, is releated to biology and resides in the fact that many proteins contain sequence stretches with either uniform (poly-A) or frequently observed motifs (cofactor-binding sites). Identification of this type of sequences, although helpful for classification of the protein in principle, is of limited use during Mascot searching, since it will not discriminate between the different protein candidates of the respective class.
Mascot assisted MS/MS-ion searches using fragmentation spectra mass lists constitutes an alternative and convienent approach frequently yielding high Mowse Scores - at least in cases where complete genetic sequence information for the organism under investigation has been made available. Furthermore, the same constraints apply as if denovo sequencing would have been performed.